Grâce aux avancées technologiques en matière de séquençage génétique qui ont permis d’obtenir plus rapidement et à moindre coût des résultats de meilleure qualité dans l’exploration des variations du génome humain, les scientifiques ont passé la dernière décennie à cataloguer frénétiquement les variations génétiques associées aux maladies aiguës et chroniques.

Toutefois, l’identification des gènes en cause n’est que la première étape. Pour que cette information ait une incidence sur le traitement ou la prévention des maladies, il faut savoir comment les variations influent sur le fonctionnement interne des cellules. Deux nouvelles études issues du laboratoire d’Alexandre Stewart, Ph.D., à l’Institut de cardiologie de l’Université d’Ottawa ont cerné des mécanismes par lesquels les variations génétiques peuvent entraîner une maladie cardiovasculaire.
La fin d’un mystère cardiovasculaire
Le tout premier facteur de risque génétique de la maladie du cœur a été découvert dans une région du génome qu’on appelle 9p21. Il entraîne une augmentation du risque de maladie du cœur pouvant atteindre 40 %, et ce, sans égard aux autres risques connus comme le taux de cholestérol, la tension artérielle et le diabète. Jusqu’à tout récemment, on ignorait encore comment 9p21 exerçait son action.
Dans un article paru dans le numéro d’octobre de la revue Circulation, des chercheurs dirigés par Alexandre Stewart, directeur du Laboratoire de recherche translationnelle en génomique cardiovasculaire, en partenariat avec la chercheuse Hsiao-Huei Chen, Ph.D., de l’Institut de recherche de l’Hôpital d’Ottawa (IRHO), révèlent que 9p21 est responsable de la croissance incontrôlée des cellules des muscles lisses dans les artères.
« Ce sont ces cellules qui déterminent la souplesse ou la rigidité des muscles [des parois vasculaires] ainsi que la quantité de sang qui passe dans les vaisseaux sanguins, explique M. Stewart. Leur prolifération dans la paroi vasculaire a pour effet de rétrécir le diamètre du vaisseau et de limiter le débit sanguin. C’est en partie ce qui se passe dans le cas de l’athérosclérose : il y a une accumulation de cholestérol dans les cellules, mais aussi une prolifération des cellules dans la paroi vasculaire. »
En temps normal, une protéine appelée TGFß modère le développement des cellules des muscles lisses dans les artères. Mais comme l’a découvert l’équipe d’Alexandre Stewart, deux variations génétiques dans la région 9p21 ont pour effet de débrider ce processus. Par une chaîne complexe d’événements moléculaires, ces variations perturbent la façon dont un facteur de transcription se fixe directement à l’ADN pour réguler l’expression des gènes.
Lorsque cela se produit, la transmission de toute une série d’instructions moléculaires échoue, ce qui empêche la TGFß de limiter la prolifération des cellules des muscles lisses dans les artères.
« Chez les personnes qui ne présentent pas la variante de risque, la TGFß empêche la prolifération des cellules. Les personnes porteuses d’une seule copie de la variante ont un contrôle limité [sur le processus]. Les personnes qui possèdent les deux copies, toutefois, perdent ce contrôle », explique M. Stewart. S’ensuit un lent et inévitable durcissement des parois artérielles qui mènera à la coronaropathie.
Un lien entre l’inflammation et la maladie du cœur
La seconde étude, également le fruit d’un partenariat avec Mme Chen et l’IRHO, a cerné un mécanisme par lequel l’inflammation des macrophages (cellules immunitaires qui éliminent les déchets et les micro-organismes indésirables dans l’organisme) contribue au dépôt de plaque sur la paroi des artères. La plaque est une marque distinctive de la coronaropathie, principale cause des crises cardiaques.
Les résultats, publiés en août dans la revue Circulation Research, laissent entrevoir une nouvelle cible potentielle de traitement pour la maladie du cœur : une voie cellulaire contrôlée par l’IRF2BP2, un gène qui régule l’activité d’autres gènes. M. Stewart et ses collègues se sont intéressés à l’IRF2BP2 dans la foulée d’études ayant observé une inflammation des macrophages lorsque ce gène est inactif.
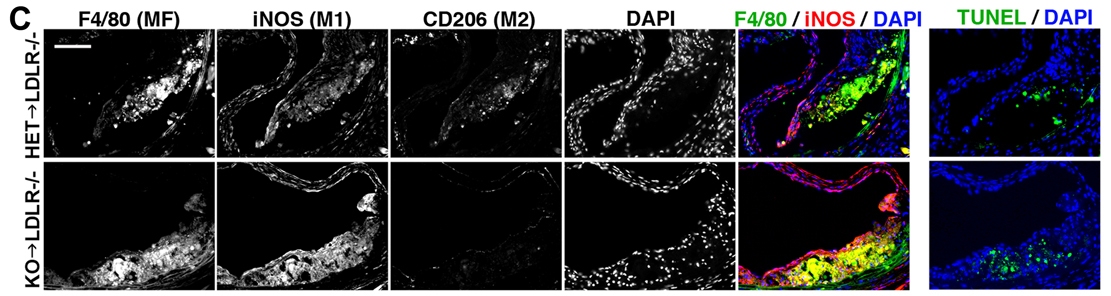
En collaboration avec le laboratoire de Hsiao-Huei Chen, neuroscientifique à l’IRHO et collaboratrice de longue date de l’Institut de cardiologie, M. Stewart et son équipe d’étudiants diplômés et de chercheurs postdoctoraux ont passé plusieurs années à créer un modèle murin dont le gène IRF2BP2 est inactivé, c’est-à-dire des souris chez qui ce gène n’est pas fonctionnel.
« Si on veut établir la fonction d’un gène dans un certain type de cellule, il faut d’abord le supprimer dans ce type de cellule et observer ce qui change, explique M. Stewart. On ne peut pas faire ce genre d’expériences avec des cultures cellulaires, car les cellules ne se comportent pas de la même façon dans un organisme. »
Les macrophages absorbent le cholestérol LDL (le « mauvais » cholestérol) de l’organisme et se mettent à le décomposer. Si ce processus est perturbé, l’inflammation gagne les macrophages, qui peuvent alors se transformer en cellules dites « spumeuses », une importante composante des dépôts de plaque caractéristiques de la coronaropathie. Les raisons et mécanismes exacts à l’origine de ce processus ne sont pas encore parfaitement compris.
Lorsque les chercheurs ont inactivé le gène IRF2BP2 des macrophages de leur modèle murin, les cellules ont produit des marqueurs d’inflammation et sont devenues plus susceptibles de se transformer en cellules spumeuses. L’équipe a ensuite greffé la moelle osseuse des souris au gène IRF2BP2 inactivé à des souris vulnérables à la coronaropathie. Sous l’effet d’une alimentation riche en gras, la moelle osseuse des greffées s’est mise à produire des macrophages enflammés. Les indicateurs de la coronaropathie se sont aussi détériorés.
Fait intéressant, ces effets négatifs étaient récessifs; autrement dit, ils n’étaient présents que chez les souris dénuées des deux copies de l’IRF2BP2. Dans le but de déterminer si cette absence modifiait aussi le risque de maladie cardiovasculaire chez l’humain, les chercheurs ont examiné l’ADN de plus de 1 000 patients atteints de coronaropathie et de 1 000 sujets témoins en bonne santé de l’Étude de génomique de l’ICUO. Chez les patients dénués des deux copies fonctionnelles de l’IRF2BP2, le risque de maladie s’est révélé substantiel : il augmentait de plus de 50 %, même après la prise en compte de facteurs de risque connus comme le tabagisme, l’hypertension et l’obésité.
Compte tenu des effets manifestement négatifs qu’entraîne son absence, la protéine IRF2BP2 constitue un domaine de recherche intéressant, estime M. Stewart. En trouvant comment maintenir un niveau élevé de cette protéine, on pourrait peut-être réduire le risque que la maladie s’aggrave. Et comme d’autres maladies peuvent aussi affecter cette protéine, un tel traitement pourrait être avantageux pour un grand nombre de personnes.