Thanks to advances in gene sequencing technology that have made better, faster, and cheaper exploration of variations in the human genome possible, scientists have spent the last decade in a mad dash to catalogue the genetic variations associated with acute and chronic diseases.

But identifying these altered genes is only the first step. For this information to play a practical role in the treatment of patients or the prevention of disease, we need to know how the variations affect the inner workings of our cells. Two new studies from the laboratory of Alexandre Stewart, PhD, at the University of Ottawa Heart Institute have pinpointed mechanisms by which genetic variations help drive cardiovascular disease.
A Cardiovascular Mystery Unraveled
The first common genetic risk factor for heart disease ever discovered was in a section of the genome called 9p21. It increases the risk of heart disease by up to 40%, regardless of other established risk factors, such as cholesterol, blood pressure and diabetes. Until now, how 9p21 exerts its effect remained a mystery.
In a paper published online in October in the journal Circulation, researchers led by Dr. Stewart, Director of the Laboratory of Translational Genomics, in partnership with Dr. Hsiao-Huei Chen of the Ottawa Hospital Research Institute (OHRI), found that 9p21 drives uncontrolled growth of smooth muscle cells in the arteries.
“It’s the arterial smooth muscle cell that controls how stiff or loose the muscles [of the blood vessel walls] are, and how much blood flows through your vessels,” explained Dr. Stewart. “When smooth muscle cells proliferate in the vessel wall, that narrows the vessel and constricts flow. That’s part of what happens in atherosclerosis: you have an accumulation of cholesterol in cells, but you also have proliferation of cells in the vessel wall.”
Normally, the development of smooth muscle cells in the arteries is kept in check by a protein called TGFß. But as Dr. Stewart’s team found, two gene variations in the 9p21 region take the brakes off this process. Through a complex molecular chain of events, these variations disrupt the way a specific transcription factor binds directly to DNA to control gene expression.
When this happens, a whole set of downstream molecular instructions fails to be passed on, and TGFß does not limit the proliferation of arterial smooth muscle cells.
“In people who don’t carry the risk variant, TGFß will keep those smooth muscle cells from proliferating. People who have one copy of the risk variant have less control [over this process]. And people who have two copies lose that control,” said Dr. Stewart. The result is a slow but inevitable stiffening of the artery walls and, eventually, coronary artery disease.
A Link Between Inflammation and Heart Disease
The second study, also done in partnership with Dr. Chen and OHRI, pinpointed a mechanism by which inflammation in macrophages—immune cells that digest waste products and invading microorganisms—contributes to plaque deposits in blood vessel walls. These plaques are the hallmark of coronary artery disease, the primary cause of heart attack.
The results, published in August in Circulation Research, point to a potential new target for treating heart disease: a cellular pathway driven by a gene called IRF2BP2, which regulates the activity of other genes. Dr. Stewart and his colleagues became interested in IRF2BP2 after studies in cells showed that macrophages become inflamed when the gene is not active.
Together with the laboratory of Hsiao-Huei Chen, PhD, a neuroscientist at OHRI and a longtime Heart Institute collaborator, Dr. Stewart and a team of graduate students and postdoctoral researchers spent several years creating a mouse model in which IRF2BP2 is “knocked out.” That is, the gene is not functional in these mice.
“If you want to know what a gene is doing in a type of cell, you need to get rid of it in that cell type and see what changes,” explained Dr. Stewart. “These aren’t experiments you can do in cell culture because those cells don’t behave the way they do in a whole organism.”
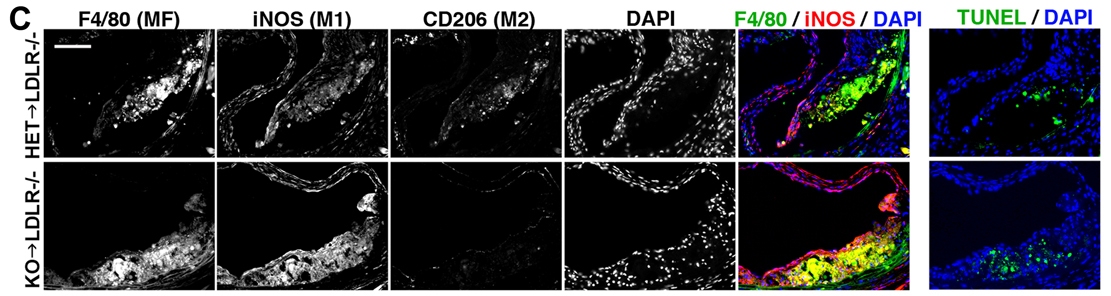
Macrophages mop up LDL cholesterol (the “bad cholesterol”) from the body and begin the process of breaking it down. If this process is disrupted, the macrophages become inflamed and can transform into what are known as foam cells, a major component of the blood vessel plaques found in coronary artery disease. How and why this process occurs is not completely understood.
When the researchers knocked out IRF2BP2 in the macrophages of their mouse model, the cells produced markers of inflammation and were more likely to form foam cells. They then transplanted bone marrow from mice lacking IRF2BP2 into other mice susceptible to coronary artery disease. When these mice were fed a high-fat diet, the marrow cells produced inflamed macrophages and indicators of coronary artery disease worsened.
Interestingly, these negative effects were recessive, meaning that they were only seen in mice lacking both copies of IRF2BP2. To see if this deletion also affects the risk of heart disease in people, the researchers examined DNA from over 1,000 patients with known coronary artery disease and 1,000 healthy controls from the Ottawa Heart Genomics Study. For patients missing both functional copies of IRF2BP2, the risk of disease turned out to be substantial: an increase of over 50%, even after adjusting for known risk factors such as smoking, hypertension and obesity.
The clearly negative effect of lacking the IRF2BP2 protein makes it an intriguing area for future research, said Dr. Stewart. Finding a way to keep the protein levels elevated might reduce the risk of worsening disease. Since other health conditions could also potentially reduce levels of the protein, an available treatment could offer benefits to a wider portion of the population.